SARS-CoV-2: Where Did It Come From and Where Does It Go Next?

Mara Aspinall, managing director, BlueStone Venture Partners; professor of the practice, biomedical diagnostics, Arizona State University
We are once again in a period of pandemic optimism — June 2021 redux. I hope this is justified, but I am reminded of the “fool me once, fool me twice” saying.
Over the past few weeks there have been four papers that examine viral infection dynamics between animals and humans — animal to human and back again (Zoonosis) that should provide a dose of caution in this otherwise optimistic moment.
The origin of SARS-CoV-2 has been hotly debated and politicized for the past two years. Origins matter – they define epidemic dynamics, and possible future variant directions. When we know where an epidemic comes from, we can better prepare for the next one.
Two papers from Michael Worobey’s team at University of Arizona (and collaborators) have been released that are compelling (although based on limited early pandemic sequence data). This latest research provides the most compelling evidence yet that the virus originated from the Wuhan Wet Market (not the Wuhan Virology Institute). One examines the genetics (phylogenetics) of the earliest cases, the other, their geographic spread.
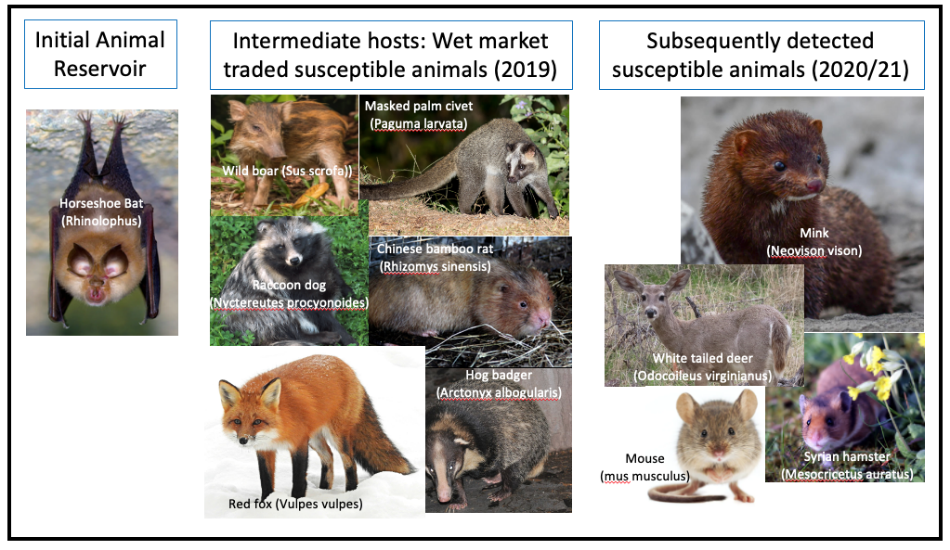
The bottom line: the first cases clearly occurred in the wet market, presumptively via intermediate hosts known to be traded there, from a long-term animal reservoir in wild Horseshoe bats.
We have known for some time that human to animal outbreaks of SARS-CoV-2 have occurred: e.g. Danish farmed mink in 2020, and in 2022, domestic hamsters in Hong Kong. A paper from Canada documents the first (and so far, only) completed loop (human to deer and back to human). A very intriguing paper from the Journal of Genetics and Genomics in December 2021 makes the case that Omicron’s enormous number of mutations was caused by a long period evolving in mice before jumping back to humans.
Very big and surprising variant jumps can occur when a virus completes a human to animal and back to human roundtrip. All four of these papers underline the importance of animal susceptibility and surveillance if we are to understand which novel variants may evolve in humans after the Omicron wave.
Of course, all the hypotheses presented, however compelling, are probabilistic based on currently available data – as new evidence emerges these can change. Nevertheless, we are at a point of knowing a lot more than we did just one year ago.
But before digging in and summarizing conclusions and implications, there are five virus principles worth remembering (see Viral Phylodynamics for details and examples):
- the mutations that create novel variants occur regularly and randomly on a virus-specific clock. Generally, the bigger a pathogen’s genome the slower they happen;
- variant evolution only happens in small steps. If jumps look big, it is only because we missed the “middle of the movie”. Both Delta and Omicron are significantly different from their ancestral variants;
- every host species has a unique mutational and immunological “evolutionary fingerprint” creating a pattern that can be traced to a specific species;
- fitness creates winners: increased transmissibility is the key to relative fitness early on in a fast-growing epidemic in an immune naïve population (i.e. SARS-CoV-2 2020-21) but immune evasion becomes dominant over time as immunity adapts (i.e. SARS-CoV-2 in 2022, when prior infection, vaccination, therapies are more widespread);
- there is no general fitness incentive for breakthrough infections to become less severe over time (e.g. HIV, Polio, Smallpox, Measles, Hepatitis C are as pathological as ever), except when a disease has an extremely high and rapid death rate (e.g. Bubonic plague, Ebola, Rabies). In those cases, new outbreaks require re-emergence from resistant species or individuals.
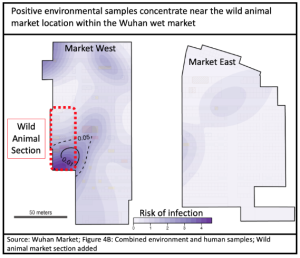
Geography is compelling that animal to human cross-over happened initially in the Wuhan Wet Market. Early cases were strongly concentrated in and around it, and primarily within the section dedicated to live animals.
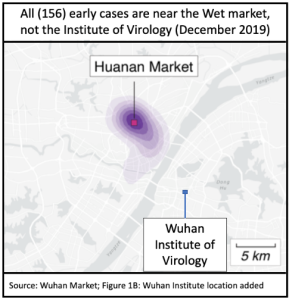
The Viral Institute lab leak conspiracy theory was initially supported by a now-debunked false claim that the furin cleavage site of the virus genome showed signs of human engineering. Beyond that, there were two popular scenarios: a lab accident infected one or a few workers; or a more extreme version in which the release was intentional.
The latter scenario is simply ludicrous. No sound malevolent plan would involve release of a virus in the same city or country where it had been developed. The former, though is certainly possible — accidents do occur. If that had happened, the geographic infection pattern would be very different — highly centered around the few exposed individuals and those who care for them. That was the case in an Ebola-like MVD virus lab contamination event in Frankfurt and Marburg in 1967. The reluctance of the Institute to release detailed records is an unforced error that has hampered disposing of this scenario conclusively, but there is simply no positive evidence for it.
Genomic (phylogenetic) evidence is a more complex, but equally compelling, argument for wet market origins. The paper concludes that at least five separate animal-to-human viral introductions likely occurred, of which two became established (lineage A and B) while 3 (or more) failed.
A consistent challenge to the wet market theory is that no specific intermediate host animal has yet been found to be the bridge from wild bats to the wet market to humans.
At first, the leading suspect was the Pangolin, but a China CDC investigation found no Pangolins had been present in the market in late 2019.
China CDC launched an extensive testing program in January and February 2020, the full details of which have only recently been published: 457 samples from 188 animals of 18 species underwent RTqPCR testing; followed by a further 80,000 animal samples from across China. No SARS-CoV-2 was found in any of them. Surfaces in and around the market were swabbed for virus, and 73 of 923 environmental samples reported positive. 7 of these were subsequently sequenced, revealing that all were from the human contamination (earliest clinical cases of Wuhan-Hu-1), therefore shedding no light on the “missing intermediate host” mystery.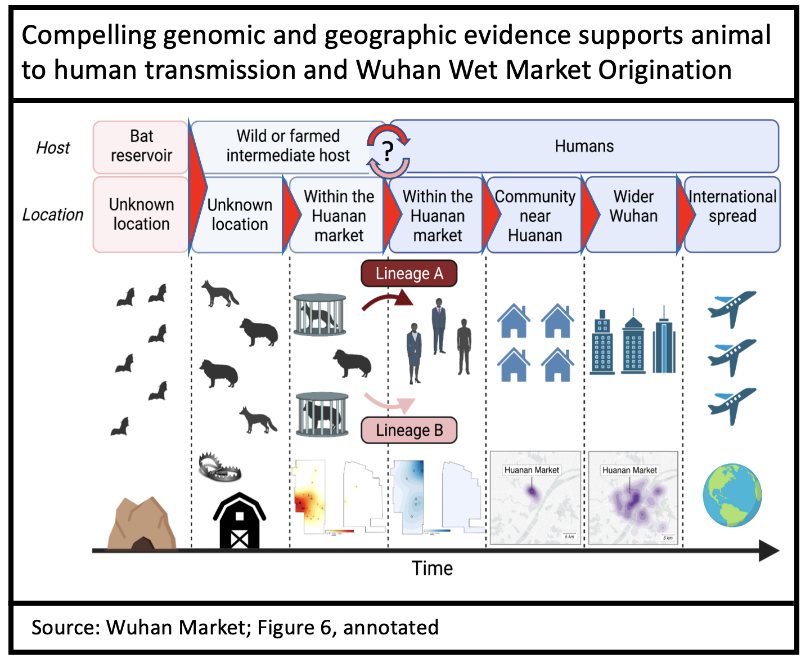
Testing techniques in 2020 were primitive by 2022 standards (and methods still not disclosed) and this likely led to this critical lack of evidence. Knowledge of the virus was very limited at the time, and testing was performed too late to have a chance to detect crossover events that must have happened months earlier, back in November and December 2019.
The most “likely to transmit” market animals were long gone by the time testing was done. The chain of transmission was broken by the market closing Jan. 1, 2020, no live animals were available to test; no serology that would have detected past infection was performed.
In a wider nationwide program (80,000 tests), the animals tested were from what we now know to be from uninfected regions; animals not then susceptible to the SARS-CoV-2 variant circulating at the time (e.g. chicken, cattle); and/or of animals butchered before the emergence of SARS-CoV-2.
Every species (including humans) that any virus inhabits presents unique mutational and immune pressures (a species-specific mutational fingerprint). All virus, e.g. SARS-CoV-2, then develop variants consistent with these unique pressures. When the virus then reappears back in humans, large mutational jumps appear to have occurred, but only because all the intermediate small steps were hidden out of sight in an animal host.
This pattern of repeated jumps between animals and humans is very similar to what happened in prior outbreaks of SARS-CoV-1 and MERS-CoV. More stable established virus types (e.g. influenza) show an incremental year-to-year evolutionary pattern, interspersed by less frequent big mutational jumps caused by cross-over from non-human avian or swine sources.
We can expect more and more of these cross-over events (animal to human) in the future since humans, not bats, are now the largest animal reservoir of SARS-CoV-2 and are frequently in contact with susceptible wild and domesticated animals. As of January 2022, 29 different species have been found to be infected with a human form of SARS-CoV-2. For example, 40 percent of free range deer tested in Michigan in 2021 were found infected with human SARS-CoV-2.
Of course, this works both ways: mouse was immune to the initial Wuhan strain, but as SARS-CoV-2 evolved in humans, a strain that could infect mouse (Beta) emerged in early 2021. Beta made only limited headway in humans, but in wild mouse a hidden epidemic occurred, during which Omicron was likely incubated: the types of mutations seen in initial Omicron (B.1.1.529) bear a mouse fingerprint on their evolution.
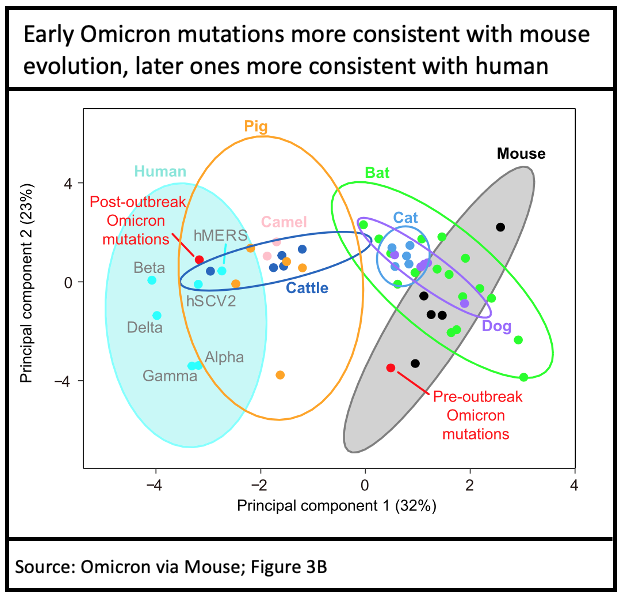
This is one hypothesis that could account for the enormous difference between Omicron and the prior human variant, Delta. Only the initial (B.1.1.529) Omicron has a mouse fingerprint, subsequent mutations to Omicron (BA.1, BA.2, BA.3) are consistent with typical in-human evolution, as expected.
There are other hypotheses for the novel and extensive mutational profile of Omicron, but all require a sustained period (3-6 months) of hidden mutational evolution. Three primary possibilities: a different non-human host; a single human host with long term chronic SARS-CoV-2 infection (e.g. an immunocompromised individual); or an isolated community of humans where the virus could mutate unobserved.
To conclude, we have only a limited and hence inadequate history of the earliest SARS-CoV-2/human relationship to rely upon to predict the future. The one thing we know for sure is that SARS-CoV-2 is not yet finished with us.
We must remember that this is the third time at bat for the virus. But unlike baseball, there is no “three strikes and you’re out” rule.
The Achilles heel of SARS-CoV-1 in 2002-2003 was early detection of fever concurrent with transmissibility. A decade later; MERS-CoV had a high fatality rate but only limited transmissibility in humans and animals (mostly camels).
SARS-CoV-2 hit a winning formula: hidden early transmissibility, now universal human exposure, and widespread non-human susceptibility.
The 4,000 year history of human airborne disease transmission is one of repeated “surprises” encountering ignorance and confusion. After the past two years we have developed extraordinary (although of course incomplete) knowledge of disease processes, genomic surveillance, physical protections, diagnostics, vaccination, and therapeutics. We need to concentrate our resources on preparedness plans that reflect our growing knowledge of this wily virus.
Ignorance is no longer a valid excuse. We must not allow this foundation to erode through wishful thinking and neglect.
You may also like

